Уважаемые пациенты! С 27 апреля клиника ЦИР на Новокузнецкой НЕ РАБОТАЕТ. Подробнее...
График. Онлайн-консультации. Оплата анализов онлайн. Анализы в другом городе.
Особенностью многих вариантных генов является то, что они могут долгое время никак себя не проявлять. Патологические симптомы могут возникнуть при дополнительных условиях (особенности питания, беременность, прием лекарств, образ жизни и т. д.). Выяснение этих дополнительных условий помогает эффективно предотвращать развитие заболеваний и их осложнений у носителей вариантных генов.
Тромбофилией называется склонность к развитию тромбов (кровяных сгустков). Тромбофилия может быть угрожающим жизни состоянием, если тромб запирает кровоток. Тромбофилия может быть наследственным нарушением, но может быть связана и с внешними причинами, такими как хирургические операции, ожирение, беременность, использование гормональных контрацептивов, антифосфолипидный синдром, повышение уровня гомоцистеина или долгий период неподвижности. Врачи подозревают наличие тромбофилии у пациентов, имевших тромбозы в прошлом, или у родственников которых были случаи тромбозов, инсультов, инфарктов в молодом возрасте (до 40 — 50 лет). Однако у многих людей с тромбофилией нет никаких симптомов, или эти симптомы проходят незамеченными, поскольку тенденция к тромбофилии недостаточно сильно выражена. Исследования последних лет показали, что наличие тромбофилии сопряжено с повышенным риском развития осложнений беременности (привычное невынашивание, плацентарная недостаточность, задержка роста плода, поздний токсикоз (гестоз)). К числу генных маркеров наследственных тромбофилий относятся мутация метилентетрагидрофолатредуктазы, лейденская мутация и мутация гена протромбина G20210A.
Исследования последних лет показали, что у пациенток с привычным невынашиванием беременности часто обнаруживаются один или несколько генетических маркеров тромбофилии. Например, в одном из исследований было обнаружено наличие лейденской мутации у 19% пациенток с невынашиванием беременности, тогда как в контрольной группе лейденская мутация была обнаружена только у 4% женщин.
Изучение MTHFR началось в 1970-е годы, когда Кутцбах и Стокстад выделили этот фермент. Исследования выявили связь наследственного дефицита этого фермента с нарушениями обмена гомоцистеина. Примерно в те же годы было показано, что повышение уровня гомоцистеина является независимым фактором риска развития сосудистых осложнений. Начались попытки выяснения генетической природы дефицита MTHFR. Клонирование гена MTHFR в 1993 г. стало основой для определения мутаций, связанных с различными степенями дефицита данного фермента.
Фермент 5,10-метилентетрагидрофолат-редуктаза относится к группе флавопротеинов и состоит из двух одинаковых субъединиц с молекулярной массой около 70 кДа. MTHFR является ключевым ферментом фолатного цикла. Фолат и фолиевая кислота (синтетический витамин, отсутсвующий в естественных продуктах) являются двумя формами семейства веществ, связанных с птероилглютаминовой кислотой (ПтеГлу). Эта кислота является сложной молекулой, состоящей из птероидной кислоты и одного (моноглютаматы) или нескольких (до 9, полиглютаматы) остатков глютаминовой кислоты (см. рис. 1). Пища, особенно свежая зелень, печень, дрожжи и некоторые фрукты в основном содержат восстановленные полиглютаматы, которые должны быть гидролизованы с помощью фермента птероилполиглютамат-гидролазы до моноглютамата, чтобы они могли быть абсорбированы в проксимальном отделе тонкого кишечника. После всасывания фолат-моноглютамат быстро восстанавливается до тетрагидрофолата, поскольку только восстановленные формы фолата обладают биологической активностью. После метилирования фолаты поступают в кровь в виде 5-метилтетрагидрофолата. Кроме пищи, постоянное поступление 5-метилтетрагидрофолата обеспечивается кишечно-печеночным циклом: птерил-моноглютамат всасывается из кишечника и поступает в печень, где он восстанавливается и метилируется до 5-метилтетрагидрофолата. Образовавшийся 5-метилтетрагидрофолат выделяется с желчью в кишечник, где он затем всасывается и разносится с кровью по всему организму.
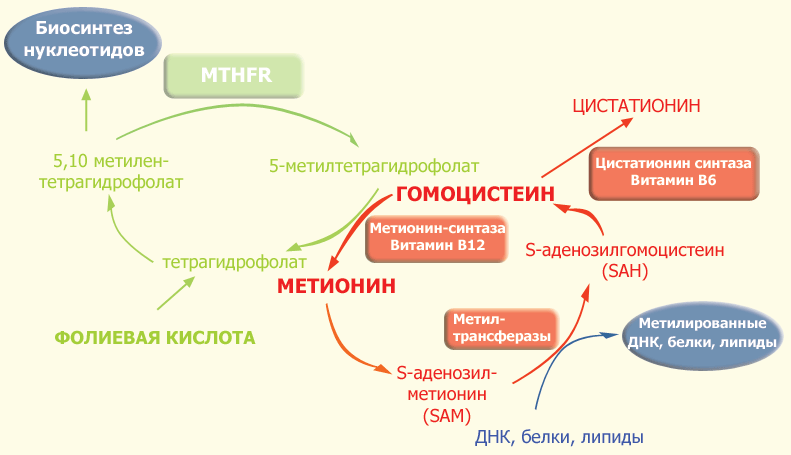
Рис. 1. Фолатный цикл и цикл метионина.
В ткани поступление 5-метилтетрагидрофолата внутрь клетки происходит с помощью эндоцитоза при участии специфических фолатных рецепторов. Описаны три изоформы фолатных рецепторов. Внутри клетки 5-метилтетрагидрофолат служит донором метильных групп и основным источником тетрагидрофолата. Последний выступает в качестве акцептора большого числа моноуглеродных групп, превращаясь в разные виды фолатов, служащих в свою очередь специфическими коферментами в целом ряде внутриклеточных реакций. К ним относятся 5-формилтетрагидрофолат (фолиниевая кислота, лейковорин), 10-формилтетрагидрофолат и 5,10-метилентетрагидрофолат.
Одной из реакций, требующих наличия 5,10-метилентетрагидрофолата и 5-метилтетрагидрофолата, является синтез метионина из гомоцистеина (путь реметилирования в обмене гомоцистеина). В этой реакции MTHFR играет ключевую роль, восстанавливая 5,10-метилентетрагидрофолат до 5-метилтетрагидрофолата, являясь таким образом катализатором единственной внутри клетки реакции образования 5-метилтетрагидрофолата. Хотя в сыворотке и других тканевых жидкостях обнаруживаются разные формы фолатов, главной формой фолата в плазме является 5-метилтетрагидрофолат, несущий на себе метильную группу, необходимую для превращения гомоцистеина в метионин. В этой реации метильная группа вначале переносится на коб(I)аламин (форма витамина B12), превращая его в метилкобаламин, который затем отдает метильную группу гомоцистеину, образуя метионин с помощью фермента метионин-синтазы. Однако в некоторых случаях коб(I)аламин может окисляться в коб(II)аламин, что приводит к подавлению метионин-синтазы. Для поддержания активности фермента необходимо восстановительное метилирование с помощью фермента метионин-синтаза-редуктазы.
Поскольку кобаламин (витамин B12) служит акцептором метильной группы 5-метилтетрагидрофолата, дефицит этого витамина приводит к "ловушке для фолата". Это тупиковый путь метаболизма, поскольку метилтетрагидрофолат не может при этом восстанавливаться до тетрагидрофолата и возвращаться в фолатный пул. Неспособность регенирировать метионин приводит к истощению запаса метионина и выбросу в кровь избытка гомоцистеина.
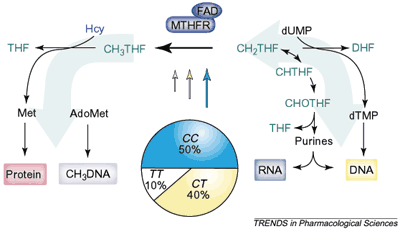
Рис. 2 C677T-полиморфизм 5,10 метилентетрагидрофолат-редуктазы (MTHFR) влияет на распределение соединений фолиевой кислоты (выделены зеленым цветом), используемых для синтеза ДНК и РНК, и 5-метилтетрагидрофолата, необходимого для реметилирования гомоцистеина (Hсy), а, значит, — для синтеза белка. Секторная диаграмма показывает распределение генотипов, типичное для европейских популяций, а размеры стрелок показывают относительную ферментную активность MTHFR.
Ген MTHFR у человека расположен на коротком плече первой хромосомы (1p36.3). Длина всего кодирующего региона составляет около 1980 п.н. с расчетной молекулярной массой продукта 74,6 кДа. Последовательность аминокислот эволюционно сохранна, поскольку имеется 90% гомологии с мышиным полипептидом MTHFR. Была расшифрована и геномная организация гена. Он состоит из 11 экзонов длиной от 102 до 432 п.н. и интронов длиной от 250 до 1500 п.н., за исключением одного интрона длиной 4200 п.н.
Описано две разновидности гена MTHFR. Наиболее изученной является вариант, в котором нуклеотид цитозин (C) в позиции 677, относящейся к 4-му экзону, заменен на тимидин (T), что приводит к замене аминокислотного остатка аланина на остаток валина в сайте связывания фолата. Такой полиморфизм MTHR обозначается как мутация C677T. У лиц, гомозиготных по данной мутации, отмечается термолабильность MTHFR и снижение активности фермента примерно до 35% от среднего значения. Кроме того, у лиц, гомозиготных по данной мутации, отмечается нарушенное распределение фолатов в эритроцитах, выражающееся в накоплении формильных полиглютаматов тетраглютамата и метилированных дериватов тетрагидрофолата. Наличие этой мутации сопровождается повышением уровня гомоцистеина в крови.
Другим вариантом полиморфизма гена MTHFR является замена нуклеотида аденина (A) на цитозин (C) в позиции 1298. Это приводит к замене остатка глутамина на остаток аланина в регуляторном домене фермента, что сопровождается небольшим снижением активности. У лиц, гомозиготных по мутации А1298C, отмечается снижение активности MTHFR примерно до 60% от нормы. Предполагается, что снижение активности фермента связано с изменением регуляции фермента его ингибитором S-аденозилметионином.
В отличие от полиморфизма C677T, гетерозиготность и гомозиготность по мутации А1298C не сопровождается ни повышением концентрации общего гомоцистеина, ни снижением уровня фолата в плазме. Однако комбинация гетерозиготности аллелей 677T и 1298C сопровождается не только снижением активности фермента, но и повышением концентрации гомоцистеина в плазме и снижением уровня фолата, как это бывает при гомозиготности 677T.
Диагностика гомо- и гетерозиготности по аллелям 677T и 1298C производится методом полимеразной цепной реакции (ПЦР).
Аллель 677T широко распространен в популяции. Частота гомозиготности составляет около 10—12%, а гетерозиготности — около 40% у европейской расы. Существуют значительные межрасовые и межэтнические различия. Чаще всего ген встречается у европейцев, реже всего — у чернокожих африканцев, и аборигенов Австралии и Шри-Ланки.
В Европе наименьшая частота аллеля 677T обнаруживается у скандинавов, а наибольшая — у южан (жителей Средиземноморья). Независимо от региона, наличие аллеля 677T связано с повышением уровня гомоцистеина плазмы, у гомозигот это повышение выражено в гораздо большей степени, чем у гетерозигот.
Высокая частота аллеля 677T предполагает, что носители данной мутации могли иметь определенные преимущества в естественном отборе. Существует гипотеза, что во время голода снижение активности MTHFR приводит к снижению реметилирования гомоцистеина, и таким образом сберегает моноуглеродные радикалы тетрагидрофолатного метаболизма для жизненно важного синтеза ДНК и РНК. Согласно другой гипотезе, носители мутантного аллеля имеют меньшую вероятность заболевания раком толстой кишки, в результате чего частота мутации в популяции может постепенно возрастать.
Мутация 677T предрасполагает к развитию умеренной гипергомоцистеинемии, особенно на фоне снижения фолатного статуса. Данное взаимодействие генетической предрасположенности и особенностей питания приводит к повышению риска развития дефектов нервной трубки у плода. Исследования выявили повышенную частоту обнаружения аллеля 677T среди матерей, отцов и детей при обнаружении дефекта нервной трубки у плода. Была обнаружена корреляция частоты аллеля 677T в популяции с частотой дефектов нервной трубки.
В настоящее время связь дефектов нервной трубки у плода с гомозиготностью матери по аллелю 677T считается доказанной. Однако не всегда развитие дефектов нервной трубки, обусловленное низким фолатным статусом у беременных, связано с аллелем 677T, что указывает на важность адекватного поступления фолиевой кислоты в организм во время беременности. Сочетание аллеля 677T с низким фолатным статусом сопряжено с большим риском развития дефектов нервной трубки, чем наличие каждого из этих двух факторов по отдельности.
Женщины с генотипом 677TT склонны к развитию витаминодефицитного состояния по фолиевой кислоте. У небеременных женщин, гомозиготных по данному аллелю, фолатный дефицит может обнаруживаться только в эритроцитах, а уровень фолатов в плазме может быть не нарушен. Однако во время беременности у гомозиготных женщин отмечается снижение концентрации фолатов не только внутри эритроцитов, но и в плазме крови.
Исследования показали повышение риска развития нефропатии у беременных с сосудистыми заболеваниями. Это хорошо согласуется с данными о влиянии высоких концентраций гомоцистеина в крови с риском развития нефропатии у беременных. Кроме того было показано, что концентрация гомоцистеина в крови коррелирует с концентрацией фибронектина в клетках, что указывает на важную роль гомоцистеина в развитии эндотелиальной дисфункции при беременности. Повышение частоты аллеля 677T было отмечено не только при позднем токсикозе (гестозе), но и при других осложнениях беременности (отслойке плаценты, задержке роста плода, антенатальной смерти плода). Сочетание аллеля 677T с другими факторами риска приводит к повышению риска раннего выкидыша. Добавление фолиевой кислоты в рацион значительно снижает риск развития осложнений беременности. Профилактическое значение добавления фолиевой кислоты в рацион особенно выражено при наличии гипергомоцистеинемии.
У лиц с тяжелыми дефицитами MTHFR часто обнаруживаются психические нарушения, отвечающие на терапию фолиевой кислотой. Поэтому существует гипотеза, что аллель 677T связан с повышенным риском развития шизофрении, тяжелых депрессивных нарушений и других психозов. Однако убедительных данных за то, что аллель 677T повышает риск развития психических заболеваний, пока получено не было. Однако не исключается участие аллеля 677T в развитии психических нарушений в сочетании с другими факторами риска.
Лейденская мутация гена V фактора свертывания крови характеризуется заменой нуклеотида гуанина на нуклеотид аденин в позиции 1691. Это приводит к замене аминокислоты аргинина на аминокислоту глутамин в позиции 506 в белковой цепи, являющейся продуктом этого гена. Напомним, что каждую аминокислоту кодирует три нуклеотида ДНК, называемые кодоном. Поэтому лейденская мутация может обозначаться как G1691A (гуанин на аденин); Arg506Gln (аргинин на глютамин) или R506Q (R — однобуквенное обозначение аргинина, Q — однобуквенное обозначение глютамина). Все три обозначения являются синонимами одной и той же мутации.
Ген V фактора свертывания находится на первой хромосоме. Мутация наследуется по аутосомно-доминантному принципу. Это означает, что повышенная склонность к тромбозам, возникающая при замене R506Q, проявляется при наличии измененного гена только на одной первой хромосоме (на другой первой хромосоме ген фактора V не изменен). Такое состояние называется гетерозиготностью. Лейденская мутация достаточно широко распространена в популяции. Гетерозиготными носителями является в среднем 4—6% европейского населения. Случаи гомозиготного носительства лейденской мутации (измененный ген на обеих первых хромосомах) в популяции втречаются крайне редко.
Мутация названа лейденской в связи с тем, что Лейденская группа исследования тромбофилии впервые расшифровала генную природу нарушений свертывания крови, возникающих при данной мутации. Это произошло в 1993 г.
Фактор V свертывания крови является высокомолекулярным белком, входящим в состав протромбиназного комплекса. Протромбиназный комплекс возникает при активации свертывания крови по внешнему или внутреннему пути и состоит из активированного фактора X (обозначается как Xa), активированного фактора V (обозначается как Va) и ионов кальция, связанных с фосфолипидными (ФЛ) мембранами (как правило, это мембраны тромбоцитов). Функция протромбиназного комплекса заключается в отщеплении от молекулы протромбина пептидных фрагментов, превращающем протромбин в тромбин (фермент, осуществляющий полимеризацию фибрина из фибриногена). Фибрин является конечным продуктом свертывания крови. Ферментом, расщепляющим протромбин в протромбиназном комплексе, является фактор Xa, однако без участия фактора V эта реакция протекает очень медленно. Активированный фактор V, соединяясь с Xa на фосфолипидной поверхности, ускоряет реакцию образования тромбина в десятки тысяч раз. (см. рис. 3).
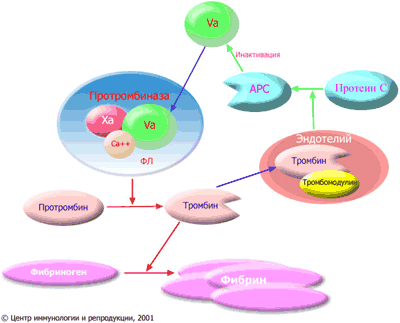
Рис. 3. Образование фибрина и инактивация протромбиназного комплекса активированным протеином С. Красными стрелками показаны реакции коагуляции, зелеными стрелками — реакции антикоагуляционной системы. Xa: активированный фактор X; Va: активированный фактор V; Ca++: ионы кальция; ФЛ: фосфолипиды; APC: активированный протеин C (activated protein C). Тромбин, образующийся при активации системы свертывания крови, вымывается из сгустка и поступает в кровь. На мембране клеток эндотелия (выстилки сосудов) он соединяется с белком тромбомодулином и теряет способность участвовать в образовании фибрина. Комплекс тромбин/тромбомодулин активирует протеин C, отщепляя от него участок молекулы. Активированный протеин С расщепляет активированный фактор V и тем самым препятствует неуправляемому расширению процесса свертывания крови. Неспособность протеина С инактивировать протромбиназный комплекс называется резистентностью к APC. Такое состояние характеризуется повышенной склонностью к тромбозам. Самой частой причиной APC-резистентности является лейденская мутация, когда активированный фактор V становится устойчивым к расщеплению активированным протеином C в результате замены аргинина на глютамин в позиции 506. Расщепление активированного фактора V происходит не в составе протромбиназного комплекса, а в несвязанном с фактором Xa виде. Таким образом, активированный протеин C не действует на готовый протромбиназный комплекс, но препятствует его образованию и регенерации. Поэтому активированный протеин С играет огромную роль в препятствии тромбообразованию в неповрежденных сосудах, в меньшей степени влияя на тромб, формирующийся в месте кровотечения.
Особенностью системы свертывания крови является наличие большого количества реакций положительной и отрицательной обратной связи. Гармоничное сочетание всего комплекса реакций позволяет организму эффективно справиться с кровотечением и не допустить тромбирования сосудов там, где кровотечения нет. Важным звеном антикоагуляционного каскада является ограничение тромбообразования активированным протеином C (латинская буква C).
Главный фермент коагуляции - тромбин - является одним из самых загадочных и интересных белков организма. Он выполняет ферментную функцию, но также может играть роль сигнальной молекулы, участвуя в целом ряде реакций организма, связанных не только с тромбообразованием. В качестве фермента тромбин выполняет две прямо противоположные функции: образовании фибрина и остановке фибринообразования. Антикоагулянтные свойства тромбин получает, соединяясь с тромбомодулином, мембранным белком эндотелия (клеток выстилки кровеносных сосудов). Молекула тромбина при этом так меняет свою конфигурацию, что становится неспособной участвовать в реакции коагуляции, но приобретает свойство расщеплять протеин C, один из витамин-К-зависимых белков, синтезируемых в печени и постоянно находящихся в кровотоке. [В 1970-е годы исследователи, изучавшие витамин-К-зависимые белки печени, обозначили их буквами латинского алфавита. Еще одним витамин-К-зависимом протеином антикоагуляционного каскада является кофактор активированного протеина C протеин S. Работ, посвященных другим белкам этой серии (протеину Z и протеину M), в последнее время выходит мало.]
Активированный протеин C является одним из главных физиологических антикоагулянтов, ращепляющих активированные факторы свертывания V и VIII. Одной из важных причин тромбофилии является устойчивость этих факторов к разрушающему действию APC. Такое состояние называется резистентностью к APC. Главной причиной такой резистентности является лейденская мутация.
В нормальном состоянии APC инактивирует фактор V, тем самым препятствуя его включению в протромбиназный комплекс. Для инактивации фактора Va активированным протеином C необходимо наличие аргинина в позиции 506. Замена аргинина на глютамин приводит к тому, что фактор V становится устойчивым к расщеплению APC. Кроме того, инактивированный фактор V необходим для инактивации VIII фактора свертывания крови комплексом протеин С/протеин S. Поэтому недостаточное образование инактивированного фактора V приводит к тому, что образование активированного фактора X, входящего в протромбиназный комплекс, также перестает блокироваться активированным протеином C. Таким образом, в организме возникают условия, способствующие гиперактивации протромбиназного комплекса, что может приводить к развитию тромбоза.
В обычном состоянии у носителя лейденской мутации может и не быть тромбозов. Тромбозы развиваются при наличии дополнительных факторов риска: беременности, приема гормональных контрацептивов, повышения уровня гомоцистеина, мутаций MTHFR и гена протромбина, антифосфолипидных антител. Важно отметить, что гомоцистеинемия сама по себе приводит к развитию резистентности к APC, поэтому такое сочетание становится особенно опасным. Кроме того, сочетание лейденской мутации с мутацией гена протромбина G20210A встречается чаще, чем этого можно было бы ожидать при случайном распределении. Все это указывает на важность достаточно полного обследования пациента при подозрении на наличие тромбофилического состояния.
Наличие лейденской мутации повышает вероятность развития целого ряда осложнений беременности: невынашивания беременности на ранних сроках (риск повышается в 3 раза), отставания развития плода, позднего токсикоза (гестоза), фетоплацентарной недостаточности. Чаще всего у женщин с лейденской мутацией обнаруживаются тромбозы в плаценте, что и является причиной повышенного риска развития всех вышеперечисленных осложнений. Профилактикой развития этих осложнений является назначение малых доз аспирина, начинаемое еще до наступления беременности, и подкожные инъекции малых доз препаратов гепарина (нефракционированного гепарина и низкомолекулярных гепаринов). Такое лечение является безопасным для плода и позволяет резко снизить шансы неблагоприятного исхода беременности.
Одним из самых опасных осложнений гормональных контрацептивов являются тромбозы и тромбоэмболии. Оказалось, что многие женщины с такими осложнениями являются гетерозиготными носителями лейденской мутации. На фоне приема гормональных контрацептивов риск тромбозов повышается в 6—9 раз. При наличии у пациентки лейденской мутации риск развития тромбозов на фоне приема контрацептивов повышается в 30—50 раз. Поэтому некоторые авторы предлагают обследовать на наличие лейденской мутации всех женщин, принимающих гормональные контрацептивы или собирающихся их принимать.
Тромбозы являются одним из грозных осложнений послеоперационного периода. Сторонники новой генетики (геномики) предлагают обследовать на наличие лейденской мутации всех пациентов, готовящихся к большим операциям (миома матки, кесарево сечение, кисты яичников и пр.).
Недавнее исследование (Lancet 2001 Oct 13;358(9289):1238-9) показало, что у носителей лейденской мутации частота успеха подсадок зародышей при IVF (по русски "ЭКО") примерно в 2 раза выше, чем среди пациенток, не являющихся носителями данной мутации. Эти любопытные данные указывают на то, что, несмотря на повышенную вероятность развития осложнений, фертильность пациенток с лейденской мутацией (вероятность наступления беременности в каждом цикле) может быть выше. Это может быть одним из объяснений того, почему данная мутация так распространилась в популяции после своего появления около 20 тысяч лет назад. Эффективное тромбирование сосудов в месте имплантации может быть важным условием успеха самых первых этапов взаимодействия зародыша со слизистой оболочкой матки. Кстати, именно поэтому избыточная гипокоагуляция не рекомендуется в дни подсадки зародышей и в предполагаемые дни имплантации при лечении нарушений репродуктивной функции, связанных с тромбофилией.
Мутация гена протромбина G20210A характеризуется заменой нуклеотида гуанина на нуклеотид аденин в позиции 20210. Мутация была открыта Лейденской группой исследования тромбофилии в 1996 г. Особенностью данной мутацией является то, что замена нуклеотида располагается в 3.-нетранслируемом участке (участке, располагающемся в конце ДНК-последовательности гена, который не транслируется). Это означает, что нуклеотидная последовательность измененного участка не участвует в кодировании аминокислотной последовательности гена протромбина. Поэтому никаких химических изменений самого протромбина при наличии данной мутации не возникает. При наличии данной мутации обнаруживаются повышенные количества химически нормального протромбина. Уровень протромбина может быть в полтора-два раза выше, чем в норме.
Ген протромбина располагается в одиннадцатой хромосоме. Гетерозиготными носителями геня являются 2-3% представителей европейской расы. Гомозиготный вариант мутации является очень редкой находкой. Среди африканцев и представителей монголоидной расы данная мутация встречается очень редко. Мутация наследуется по аутосомно-доминантному типу. Это означает, что тромбофилия возникает даже у гетерозиготного носителя измененного гена.
При возникновении тромбозов мутация G20210A часто встречается в сочетании с лейденской мутацией. Данная мутация является фактором риска всех осложнений, связанных с лейденской мутацией (невынашивание беременности, фето-плацентарная недостаточность, внутриутробная гибель плода, гестозы, задержка развития плода, отслойка плаценты).
Тромбофилические состояния (антифосфолипидный синдром, гипергомоцистеинемия, мутации генов MTHFR, V фактора и протромбина) являются одной из важных причин невынашивания беременности и фето-плацентарной недостаточности. Вне беременности данные состояния могут быть причиной тромботических осложнений гормональных контрацептивов и хирургических операций. Мы рекомендуем проводить молекулярно-генетическое обследование в следующих случаях: